Understand SMA
SMA is a neurodegenerative disorder that—left untreated—can result in progressive muscular atrophy, and in its most severe forms, premature death1,2
Spinal muscular atrophy (SMA) is a rare genetic disease caused by the deletion or mutation of the survival motor neuron 1 (SMN1) gene. The SMN1 gene produces survival motor neuron (SMN) protein that is critical for normal function of motor neurons.
Patients with SMA have an insufficient amount of SMN protein, which leads to permanent loss of motor neurons. Untreated, SMA Type 1 is the number one genetic cause of infant death.3-5
Diagnosis and treatment choice
Hear from experts in SMA care on the cause of SMA, how ZOLGENSMA works, and what to expect during the treatment decision conversation.
Transcript
Transcript: EXPERTZ – DIAGNOSIS AND TREATMENT CHOICE
ZOLGENSMA (onasemnogene abeparvovec-xioi) is a gene therapy for the treatment of pediatric patients less than 2 years of age with spinal muscular atrophy (SMA).
ZOLGENSMA has a Boxed Warning for Serious Liver Injury and Acute Liver Failure. Please see additional Important Safety Information at the end of this video. Please see the accompanying Full Prescribing Information.
EXPERTZ: Medical Experts Series
DR SHIEH
The new diagnosis of SMA is an emergency.
DR RAO
A new SMA diagnosis is always emotional.
DR TESI ROCHA
It’s not easy for the families and the patients that come, but it’s also not easy for us.
DIAGNOSIS AND TREATMENT CHOICE
How do you manage the emotional impact of a new SMA diagnosis?
DR RAO
I think it’s both emotional for the families as well as the care provider because it’s a very charged environment when you’re giving somebody a diagnosis with a disease that can be very progressive and very disabling.
DR SHIEH
So, when we get a new diagnosis on newborn screen, we call the patient immediately and we ask them to come in within days, preferably the next day if possible, so that we can start talking about the diagnosis. It’s very important for us to get the ball rolling so that we can map out when we’re going to be able to start treatment.
DR RAO
It’s a very emotional time, and the amount of information that you want to deliver can be overwhelming.
DR TESI ROCHA
So, when we set up the clinic visit days for those new diagnosis, we make sure that we will have a lot of time to devote to them.
How do you explain SMA to a family receiving a new diagnosis?
DR SHIEH
When we’re explaining SMA nowadays is we’re explaining to a family that their child has a severe, progressive, devastating disease. But of course, this looks different than the picture that they see of their child who looks like a perfectly healthy baby. And so, it’s important for us to give them time so that we can explain and perhaps re-explain what SMA is.
DR RAO
One of the first questions is to keep it very simple and say, “What do you already know? And let’s start from there.”
DR TESI ROCHA
There’s some families that know a lot, there’s some others that this is all new to them. So, for the ones that this is all new to them, I usually use visual aids such as…I like to draw myself to understand something. So, I usually use drawing to explain anything from chromosomes to genes. And then I also work with a genetic counselor, and she usually also brings some visual aids. And I think those are very well taken by the families to understand these very complicated, abstract medical terms.
DR RAO
We start by explaining that spinal muscular atrophy is caused by the absence of the two copies of the SMN1 gene, and then there’s a backup gene called the SMN2. Now, the backup gene SMN2 tries to make protein, but it is not enough. And that’s why you see the clinical manifestations of Spinal Muscular Atrophy.
How do you talk to families about treatment options?
DR RAO
Our whole goal is to make sure that they understand as much as they can about all the therapies that are out there in terms of treating SMA. And we’re very fortunate to have three therapies. When I’m talking to parents about these three treatments, I tell them that all these three treatments increase the total SMN protein, but in different ways.
DR SHIEH
The SMN1 gene is a gene that’s primarily responsible for making the SMN protein and the SMN protein is what keeps the motor neurons alive. Our patients don’t have an SMN1 gene or that it’s significantly mutated and that the only SMN protein that their child is able to make relies on the SMN2 gene.
DR RAO
ZOLGENSMA increases it by giving back the missing SMN gene, which is the root cause of SMA.
DR SHIEH
The other two therapies work by enhancing the second SMN gene, or the SMN2 gene, by increasing its efficiency in making SMN protein. One is an oral therapy that needs to be taken daily and the other is a medication that’s injected intrathecally into the spinal fluid and that has to be injected every four months, although there is a loading period of two months.
DR TESI ROCHA
I then go over the expected outcomes. And for that, I use usually the websites from the different pharmaceutical companies that sponsor these treatments. I feel that both ZOLGENSMA and the other options are good treatments for the patients, we now have enough data to support that in terms of not only efficacy but safety.
How do you approach making a treatment decision?
DR RAO
We sit down with families, as a team, and help them make that decision in a shared decision-making model. We do impart a sense of urgency to the families too, in terms of making a decision, but armed with the full set of knowledge of what each decision means in terms of the benefits and risks.
DR TESI ROCHA
And they ultimately, are the ones making the decision. We certainly can help because I work as a little anthropologist trying to identify, okay, what is the lifestyle of this family? Would this be something that will be able to adhere in terms of follow-ups, visits to the physician?
DR SHIEH
In my experience, ZOLGENSMA is the most attractive and popular choice.
DR RAO
ZOLGENSMA is an attractive option for families because it’s a one-time treatment. They don’t have to come in repeatedly for the treatment itself.
DR TESI ROCHA
But it’s very important to emphasize that even if it’s a one-time-only patients do need to return for follow ups. It’s not one-time and done.
DR RAO
Patients need to take steroids after ZOLGENSMA treatment, and even start a day before infusion, because ZOLGENSMA has a Boxed Warning for serious liver injury and acute liver failure.
Monitor patients at baseline and for at least 3 months following ZOLGENSMA infusion.
Please Full Prescribing Information for complete pre- and post-treatment requirements.
Please see additional Important Safety Information at the end of this video.
Key signs of SMA
At birth, infants may appear normal, but can develop some of these signs as they age3,4,6:
- Muscle weakness and hypotonia
- Areflexia
- Impaired head control
- Reduced bulbar function, including impaired swallowing, feeding, and weak cry and cough
- Tongue fasciculations
- Paradoxical breathing, also known as “belly breathing,” and bell-shaped chest due to intercostal muscle weakness
- Progressive respiratory failure requiring noninvasive ventilation (NIV)
- Missed motor milestones
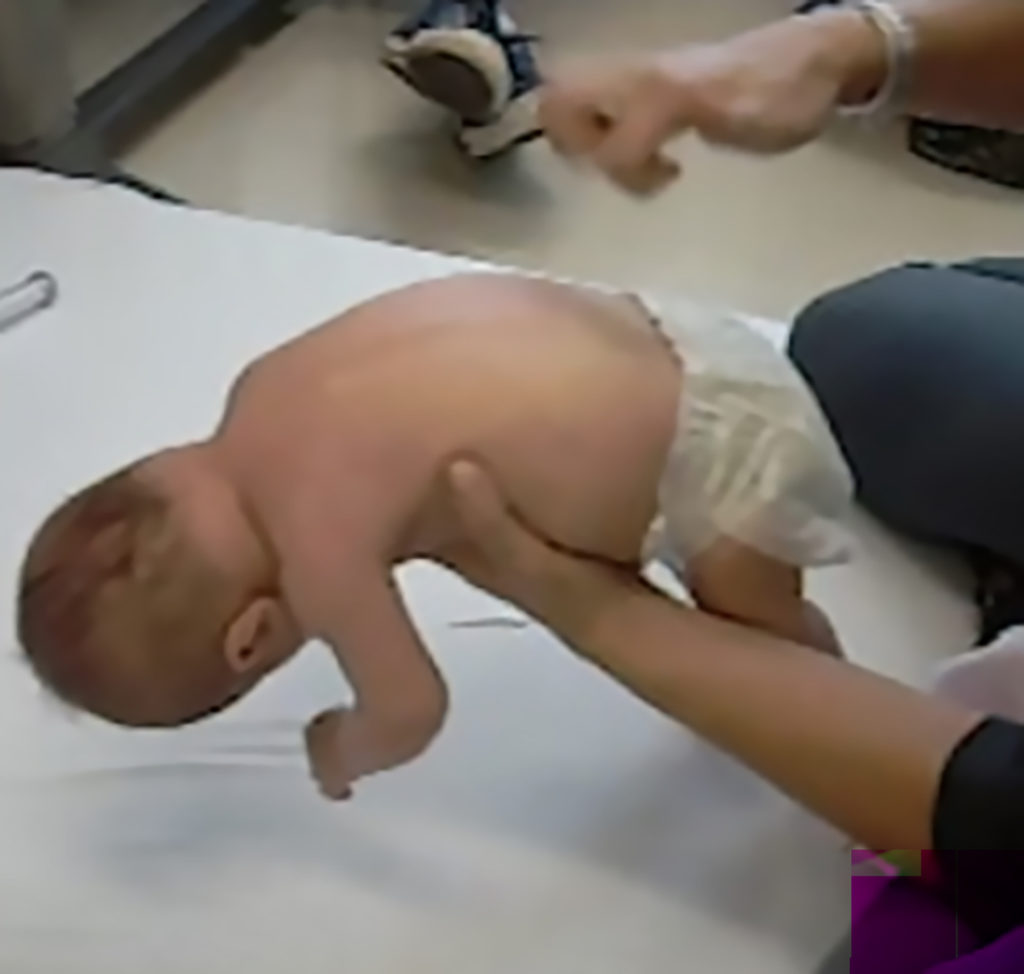
Image from Videos on File. AveXis, Inc., 2018.7
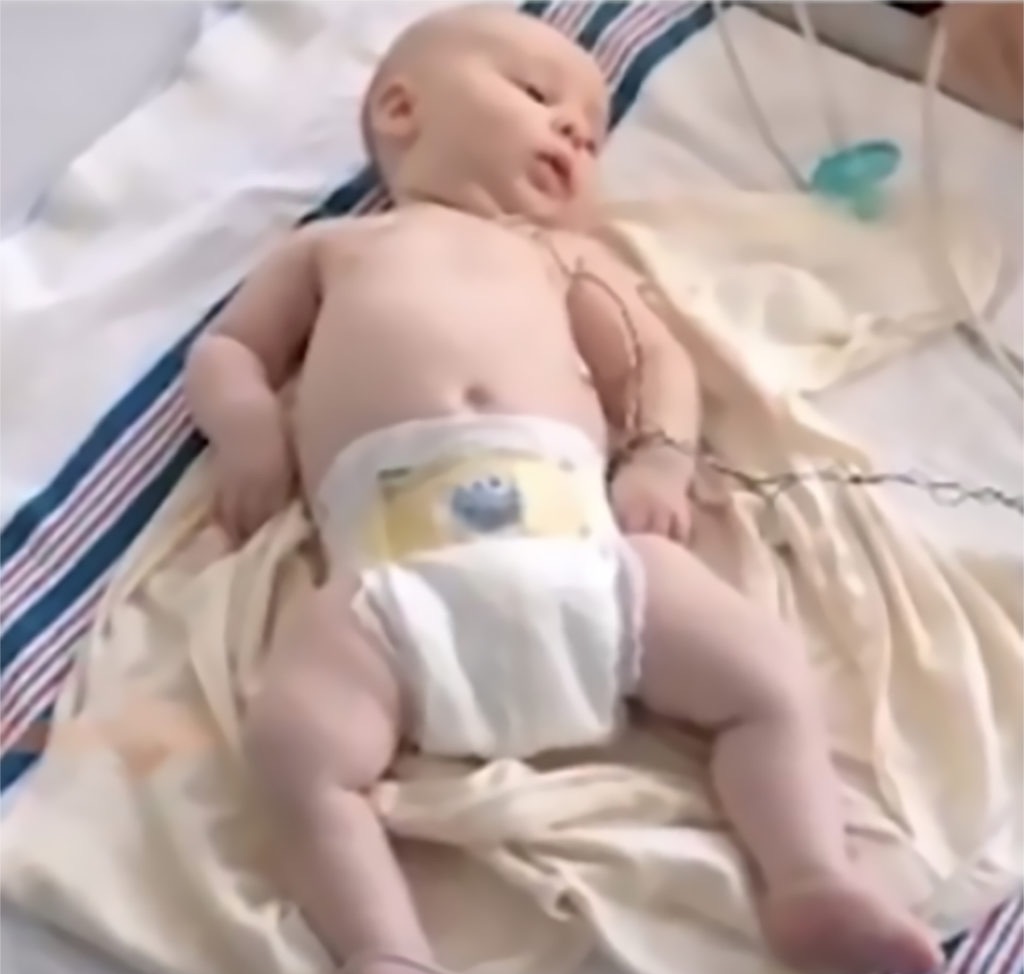
Image used with permission from CureSMA.8
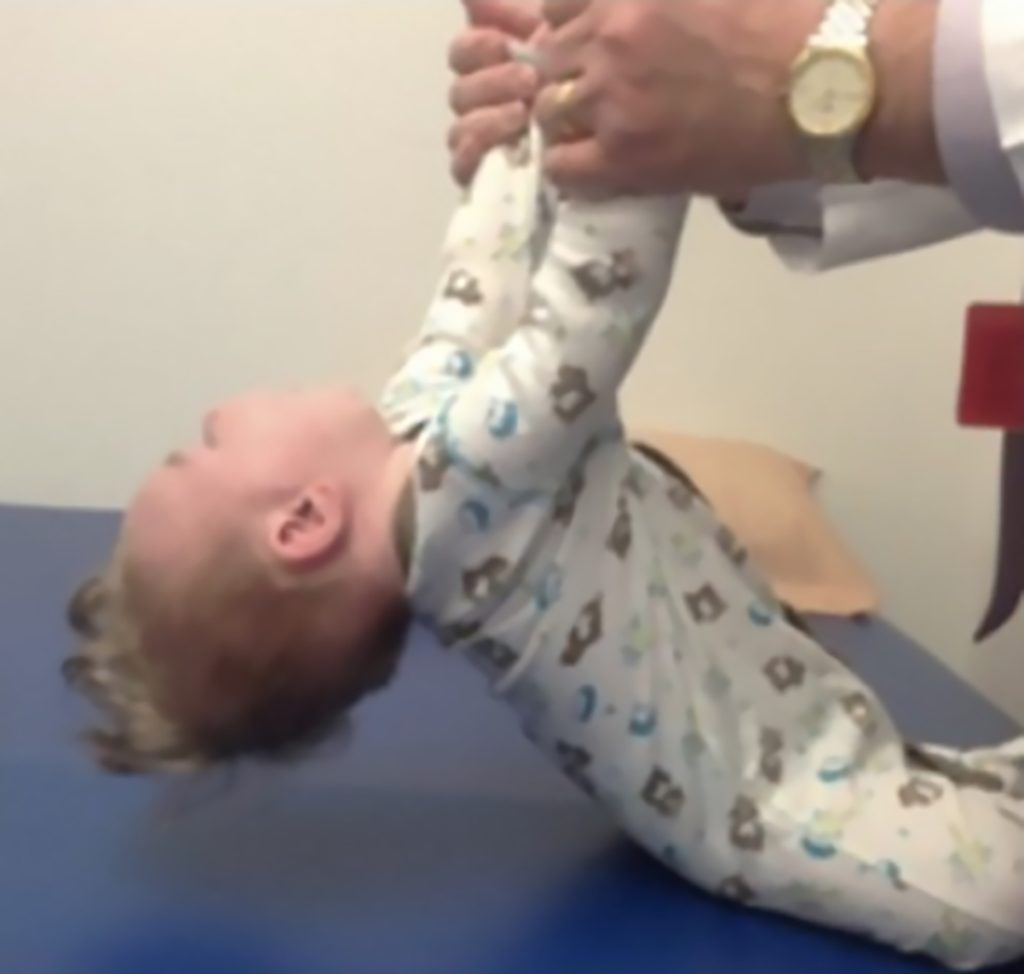
Image reprinted by permission from Elsevier. From: Sumner CJ, Paushkin S, Ko CP, eds. Spinal muscular atrophy.
Amsterdam, the Netherlands:
Elsevier; 2017.9
SMA is diagnosed with genetic testing4
SMA is classically categorized by type. Clinical features and expected outcomes are based on the natural history of untreated patients.3
If SMA is suspected, a genetic test can confirm a homozygous mutation of the SMN1 gene. Further genetic testing can determine SMN2 copy number, which is an indicator for severity. Patients with SMA rely on the SMN2 backup gene for SMN protein production; however, approximately only 10% of SMN protein produced by SMN2 is functional.3,4
| SMA Type | Type 1 | Type 2 | Type 3 |
|---|---|---|---|
| SMN15 | Nearly all patients with SMA, regardless of type, will have bi-allelic deletions or mutations of SMN1 | ||
| SMN2 copy number5,10 | 1-3 | 2-3 | 3-4 |
| Incidence rate11 | ~60% | ~27% | ~12% |
| Age of onset12 | 0-6 months | 6-18 months | >18 months |
| Maximal motor milestones achieved12 | Never achieve sitting | Sit but never walk | Stand and walk |
| Key clinical features3,4 | Severe hypotonia, respiratory insufficiency, poor feeding and head control | Scoliosis, unable to walk independently, proximal weakness | Proximal weakness, may lose ability to walk over time |
The Novartis Gene Therapies Laboratory Testing Program can support in assisting genetic testing. For more information, please contact your Novartis Gene Therapies Regional Account Associate Director or call the OneGene Program® at 1-855-441-GENE (4363)
References: 1. Talbot K, Tizzano EF. The clinical landscape for SMA in a new therapeutic era. Gene Ther. 2017;24(9):529-533. 2. Lin CW, Kalb SJ, Yeh WS. Delay in diagnosis of spinal muscular atrophy: a systematic literature review. Pediatr Neurol. 2015;53(4):293-300. 3. Kolb SJ, Kissel JT. Spinal muscular atrophy. Neurol Clin. 2015;33(4):831-846. 4. Wirth B, Karakaya M, Kye MJ, Mendoza-Ferreira N. Twenty-five years of spinal muscular atrophy research: from phenotype to genotype to therapy, and what comes next. Annu Rev Genomics Hum Genet. 2020;21:231-261. 5. Anderton RS, Mastaglia FL. Advances and challenges in developing a therapy for spinal muscular atrophy. Expert Rev Neurother. 2015;15(8):895-908. 6. Wang CH, Finkel RS, Bertini ES, et al; Participants of the International Conference on SMA Standard of Care. Consensus statement for standard of care in spinal muscular atrophy. J Child Neurol. 2007;22(8):1027-1049. 7. Data on file. AveXis, Inc. 2020. 8. Curesma.org. https://curesma.org. Accessed February 21, 2023. 9. Oskoui M, Darras BT, De Vivo DC. Chapter 1—Spinal muscular atrophy: 125 years later and on the verge of a cure. In: Sumner CJ, Paushkin S, Ko CP, eds. Spinal Muscular Atrophy. Amsterdam, the Netherlands: Elsevier; 2017. 10. Finkel RS, McDermott MP, Kaufmann P, et al. Observational study of spinal muscular atrophy type I and implications for clinical trials. Neurology. 2014;83(9):810-817. 11. Ogino S, Wilson RB, Gold B. New insights on the evolution of the SMN1 and SMN2 region: simulation and meta-analysis for allele and haplotype frequency calculations. Eur J Hum Genet. 2004;12(12):1015-1023. 12. Farrar MA, Park SB, Vucic S, et al. Emerging therapies and challenges in spinal muscular atrophy. Ann Neurol. 2017;81(3):355-368.