SMA is a neurodegenerative disorder that—left untreated—can result in progressive muscular atrophy, and in its most severe forms, premature death1,2
Spinal muscular atrophy (SMA) is a rare genetic disease caused by the deletion or mutation of the survival motor neuron 1 (SMN1) gene. The SMN1 gene produces survival motor neuron (SMN) protein that is critical for normal function of motor neurons.
Patients with SMA have an insufficient amount of SMN protein, which leads to permanent loss of motor neurons. Untreated, SMA Type 1 is the number one genetic cause of infant death.3-5

Key signs of SMA
At birth, infants may appear normal, but can develop some of these signs as they age3,4,6:
- Muscle weakness and hypotonia
- Areflexia
- Impaired head control
- Reduced bulbar function, including impaired swallowing, feeding, and weak cry and cough
- Tongue fasciculations
- Paradoxical breathing, also known as “belly breathing,” and bell-shaped chest due to intercostal muscle weakness
- Progressive respiratory failure requiring noninvasive ventilation (NIV)
- Missed motor milestones
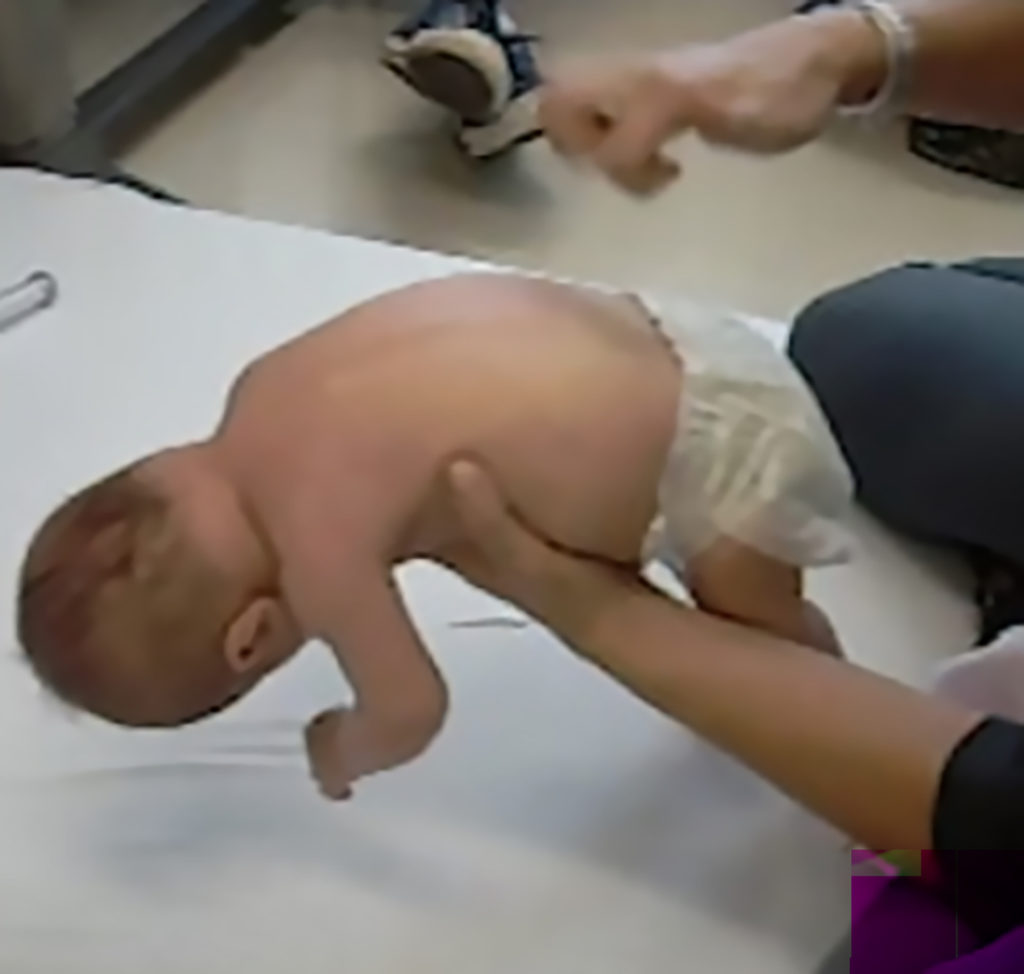
Image from Videos on File. AveXis, Inc., 2018.7
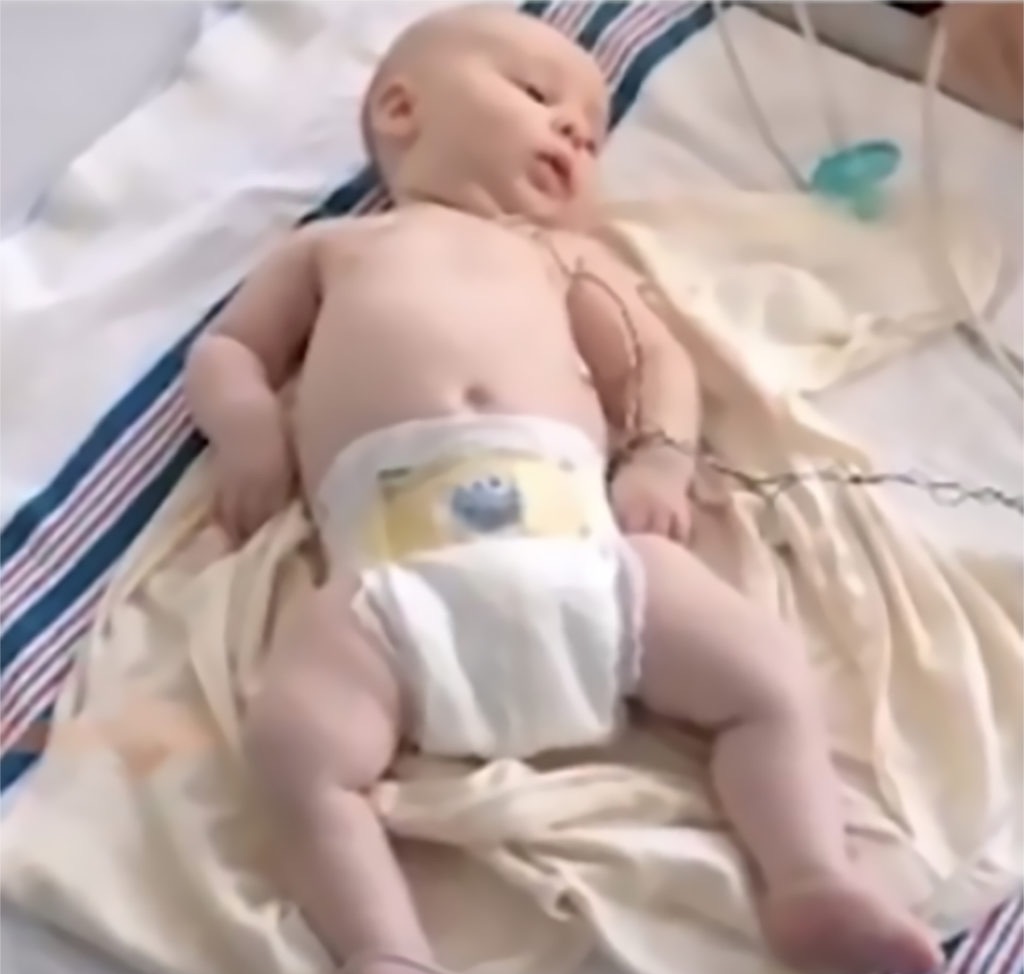
Image used with permission from CureSMA.8
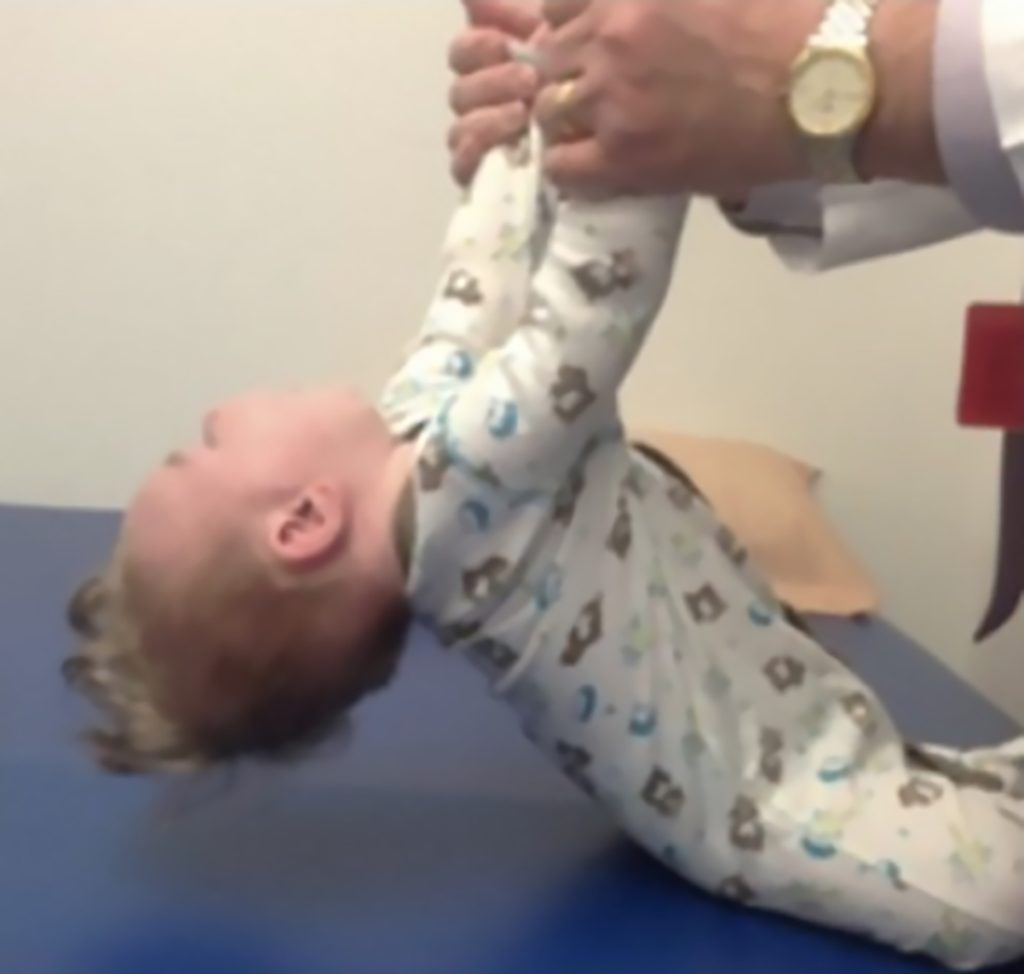
Image reprinted by permission from Elsevier. From: Sumner CJ, Paushkin S, Ko CP, eds. Spinal muscular atrophy.
Amsterdam, the Netherlands:
Elsevier; 2017.9
SMA is diagnosed with genetic testing4
SMA is classically categorized by type. Clinical features and expected outcomes are based on the natural history of untreated patients.3
If SMA is suspected, a genetic test can confirm a homozygous mutation of the SMN1 gene. Further genetic testing can determine SMN2 copy number, which is an indicator for severity. Patients with SMA rely on the SMN2 backup gene for SMN protein production; however, approximately only 10% of SMN protein produced by SMN2 is functional.3,4
| SMA Type | Type 1 | Type 2 | Type 3 |
|---|---|---|---|
| SMN15 | Nearly all patients with SMA, regardless of type, will have bi-allelic deletions or mutations of SMN1 | ||
| SMN2 copy number5,10 | 1-3 | 2-3 | 3-4 |
| Incidence rate11 | ~60% | ~27% | ~12% |
| Age of onset12 | 0-6 months | 6-18 months | >18 months |
| Maximal motor milestones achieved12 | Never achieve sitting | Sit but never walk | Stand and walk |
| Key clinical features3,4 | Severe hypotonia, respiratory insufficiency, poor feeding and head control | Scoliosis, unable to walk independently, proximal weakness | Proximal weakness, may lose ability to walk over time |
The Novartis Gene Therapies Laboratory Testing Program can support in assisting genetic testing. For more information, please contact your Novartis Gene Therapies Regional Account Associate Director or call the OneGene Program® at 1-855-441-GENE (4363)